Quer estudos de caso reais? 10 segundos para se cadastrar
Junte-se à plataforma
23 de junho de 2025 · Atualizado 7 de maio de 2026
Aproximadamente 5 minutos
Revisado por Nate Lam, Fundador e Diretor, ElendiLabs
Resposta rápida
Hong Kong classifica dispositivos médicos gerais pelo Technical Reference TR-003 do MDACS usando uma estrutura baseada em risco. A classe depende de fatores como invasividade, duração de uso, função ativa, contato com o corpo e suporte ou manutenção da vida. A classificação define as evidências e o caminho de listagem para acesso ao mercado de Hong Kong.
Classificação de Dispositivos Médicos Gerais em Hong Kong: Padrões Regulatórios e Requisitos do MDACS
O Sistema de Controle Administrativo de Dispositivos Médicos (MDACS) em Hong Kong utiliza uma estrutura de classificação baseada em risco para garantir que a supervisão regulatória seja proporcional aos riscos potenciais associados a um dispositivo médico. Para dispositivos médicos gerais, os princípios orientadores e as regras específicas para categorização estão estabelecidos na Referência Técnica TR-003, intitulada "Classificação de Dispositivos Médicos Gerais". O cumprimento deste documento é obrigatório para fabricantes e Pessoas Responsáveis Locais (LRPs), pois a classe de risco atribuída dita os procedimentos de avaliação de conformidade necessários e os requisitos gerais para o processo de listagem no MDACS.
Os princípios de classificação detalhados na TR-003 alinham-se aos padrões internacionais harmonizados, especificamente aqueles desenvolvidos pelo Fórum Internacional de Reguladores de Dispositivos Médicos (IMDRF), garantindo que o ambiente regulatório de Hong Kong permaneça consistente com as melhores práticas globais.
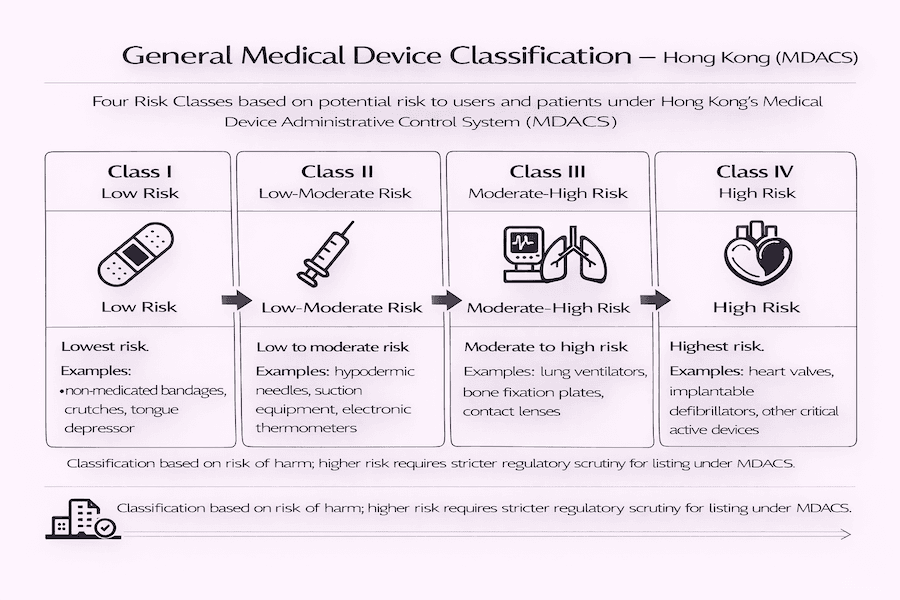
As Quatro Classes de Risco: Hierarquia de Escrutínio Regulatório
Os dispositivos médicos gerais sob a estrutura do MDACS são categorizados em quatro classes distintas com base em seu perfil de risco. À medida que o nível de risco aumenta, o grau de escrutínio regulatório e as exigências de evidências clínicas e técnicas tornam-se mais rigorosos.
- Classe I (Baixo Risco): Dispositivos de menor risco, como bandagens não medicadas, muletas ou abaixadores de língua. Geralmente, esses dispositivos não exigem listagem sob o MDACS.
- Classe II (Risco Baixo a Médio): Dispositivos de risco baixo a moderado, incluindo agulhas hipodérmicas, equipamentos de sucção e termômetros eletrônicos.
- Classe III (Risco Médio a Alto): Dispositivos considerados de risco moderado a alto; este grupo inclui ventiladores pulmonares, placas de fixação óssea e lentes de contato.
- Classe IV (Alto Risco): Dispositivos de maior risco, compreendendo itens críticos como válvulas cardíacas, desfibriladores implantáveis e outros dispositivos implantáveis ativos.
A classificação de um dispositivo determina diretamente a complexidade da solicitação de listagem e a profundidade dos dados de segurança e desempenho exigidos pela Divisão de Dispositivos Médicos (MDD).
Princípios de Classificação: Determinando a Categoria Regulatória
A TR-003 descreve critérios específicos usados para determinar a classificação de um dispositivo. Esta categorização baseia-se no uso pretendido definido pelo fabricante e no design técnico do produto. Os seguintes fatores, individualmente ou em combinação, ditam a classe de risco:
- Duração do Contato: O tempo que o dispositivo permanece em contato com o corpo (transitório, curto prazo ou longo prazo).
- Grau de Invasividade: Se o dispositivo é não invasivo, invasivo (via orifício corporal ou entrada cirúrgica) ou implantável.
- Entrega de Produtos Medicinais ou Energia: O método e a natureza da entrega de energia ou medicação ao paciente.
- Efeitos Biológicos ou Sistêmicos: Se o dispositivo se destina a ter um efeito biológico ou a ser absorvido pelo corpo.
- Uso em Combinação: A classificação pode ser influenciada pelo fato de o dispositivo ser projetado para ser usado em conjunto com outro dispositivo médico.
- Software Autônomo: Softwares que atendem à definição de dispositivo médico são classificados com base em seu uso pretendido e riscos associados, sendo normalmente categorizados como dispositivos ativos.
Aplicação das Regras de Classificação: Metodologia Regulatória
Os fabricantes devem seguir uma abordagem sistemática ao aplicar as regras de classificação definidas na TR-003:
- Avaliação Sistemática: Cada dispositivo deve ser avaliado em relação a todas as regras aplicáveis na Referência Técnica.
- O Princípio da Classe Mais Alta: Nos casos em que múltiplas regras de classificação se aplicam a um único dispositivo, deve ser adotada a regra que resulte na classificação de risco mais alta. Esta abordagem conservadora prioriza a segurança do paciente, garantindo que o dispositivo atenda aos padrões mais rigorosos aplicáveis.
- Análise Específica do Design: Embora a TR-003 forneça exemplos ilustrativos, a classificação final deve basear-se nas características técnicas específicas, indicações de uso e aplicação clínica do dispositivo individual, em vez de analogias gerais.
Responsabilidades do Fabricante e o Papel do LRP
O fabricante detém a responsabilidade primária pela classificação correta de um dispositivo médico de acordo com a TR-003. Uma fundamentação regulatória detalhada que suporte a classe de risco atribuída deve ser documentada e mantida, pois serve como um componente crítico do processo de revisão do MDACS.
A Pessoa Responsável Local (LRP) atua como o intermediário essencial para fabricantes estrangeiros em Hong Kong. O LRP garante que a solicitação de listagem reflita corretamente a classe de risco do dispositivo e esteja alinhada com as expectativas regulatórias locais. A classificação correta é a etapa fundamental do processo de registro; a classificação incorreta frequentemente resulta em atrasos significativos na aprovação, solicitações de informações adicionais ou não conformidade regulatória.
A adesão aos princípios da TR-003 garante que os dispositivos médicos no mercado de Hong Kong estejam sujeitos à supervisão apropriada, protegendo assim a saúde pública e garantindo a segurança e o desempenho das tecnologias médicas.
Precisa de Ajuda Regulatória? Experimente Nossa Plataforma
Publique suas perguntas regulatórias ou solicite orçamentos de consultores farmacêuticos verificados em todo o mundo. Conecte-se com especialistas do seu mercado.
Preços Imbatíveis
Taxas transparentes e competitivas para registro de dispositivos médicos em Hong Kong.
Farmacêutico registrado · Engenheiro de IA · Diretor, ElendiLabs
Farmacêutico registrado, engenheiro de IA, fundador da HKHAIS e especialista em SEO/GEO para o domínio farmacêutico e de dispositivos médicos.
Pergunte Qualquer Coisa
Entraremos em contato pessoalmente.
100% de taxa de resposta • Resposta em até 7 dias úteis
Precisa de Orientação Especializada?
Explore Nosso Guia Interativo de Dispositivos Médicos
Obtenha orientação passo a passo sobre as regulamentações de dispositivos médicos de Hong Kong, classificação de dispositivos e requisitos de conformidade.
Ver Guia de Dispositivos MédicosEntre em contato conosco em contact@elendilabs.com / +852 4416 5550
Artigos Relacionados
Aproximadamente 5 minutos
Relato de Eventos Adversos de Dispositivos Médicos em Hong Kong: Um Guia para LRPs
A notificação de eventos adversos é um componente crítico do Sistema de Controle Administrativo de Dispositivos Médicos de Hong Kong (MDACS), com o objetivo de melhorar a saúde e a segurança pública. Este artigo descreve os requisitos e responsabilidades das Pessoas Responsáveis Locais (LRPs) na notificação de eventos adversos relacionados a dispositivos médicos listados, com base em nossas percepções.
Aproximadamente 5 minutos
Regulamentação de dispositivos médicos em Hong Kong: guia MDACS
Guia prático sobre regulamentação de dispositivos médicos em Hong Kong, classificação MDACS, listagem, responsável local e pós-mercado.
Aproximadamente 5 minutos
Regulamentação MDACS em Hong Kong: framework e conformidade
Entenda regulamentação MDACS: framework DOH, classificação, listagem e pós-mercado.
Aproximadamente 5 minutos
Procedimentos de Listagem para Fabricantes Locais de Dispositivos Médicos em Hong Kong: Um Guia para GN-08
Este artigo detalha o processo de aplicação para fabricantes locais de dispositivos médicos que buscam ser listados sob o Sistema de Controle Administrativo de Dispositivos Médicos de Hong Kong (MDACS), conforme orientado pelo GN-08. Ele abrange elegibilidade, requisitos do sistema de gestão da qualidade e o processo de submissão, tudo baseado em nossas percepções e experiências para uma fabricação eficaz de dispositivos médicos em Hong Kong.
Aproximadamente 5 minutos
Procedimentos de Listagem para Importadores de Dispositivos Médicos em Hong Kong: Um Guia para GN-07
Para entidades que importam dispositivos médicos para Hong Kong, o Sistema de Controle Administrativo de Dispositivos Médicos (MDACS) oferece um esquema de listagem voluntária para importadores, orientado pela GN-07. Este artigo detalha a elegibilidade, os passos de aplicação e os principais requisitos para a listagem como importador de dispositivos médicos, com base em nossas percepções e experiências para uma importação eficiente de dispositivos médicos em Hong Kong.
Aproximadamente 5 minutos
Listagem de distribuidores em Hong Kong (GN-09)
Listagem de distribuidores MDACS conforme GN-09: elegibilidade, procedimentos documentados, MDIS e conformidade contínua.
Aproximadamente 5 minutos
Registro IVD em Hong Kong: listagem MDACS Classes B–D
Entenda registro IVD e listagem MDACS para IVDMD Classes B, C e D em Hong Kong.