Quer estudos de caso reais? 10 segundos para se cadastrar
Junte-se à plataforma
16 de junho de 2025
Aproximadamente 5 minutos
Revisado por Nate Lam, Fundador e Diretor, ElendiLabs
Compreendendo a Regulamentação de Dispositivos Médicos em Hong Kong: O Que Aprendemos
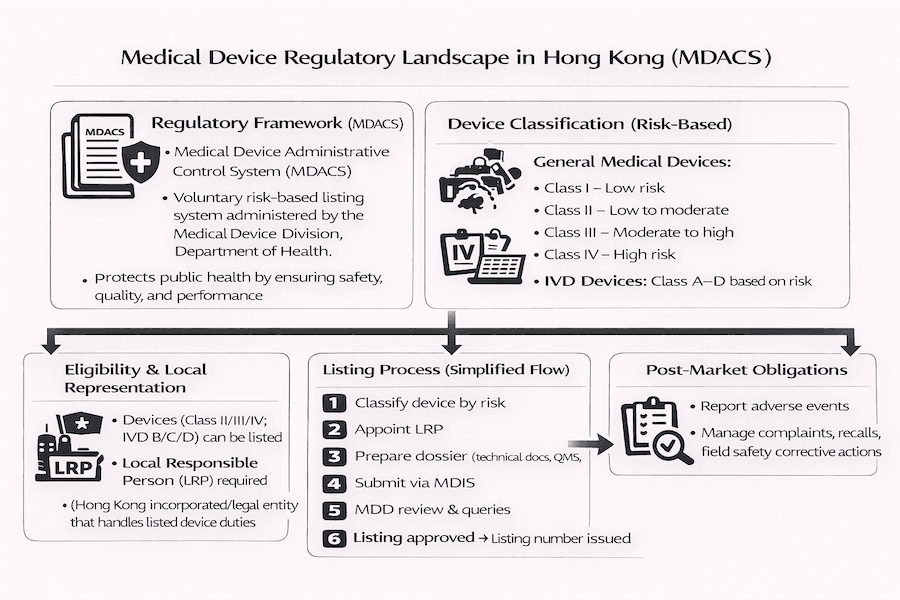
Do nosso ponto de vista, a forma como os dispositivos médicos são regulamentados em Hong Kong atualmente opera sob um sistema voluntário conhecido como Sistema de Controle Administrativo de Dispositivos Médicos (MDACS). O Departamento de Saúde estabeleceu isso, e seu objetivo principal, conforme entendemos, é proteger a saúde pública, garantindo que os dispositivos médicos sejam seguros, de alta qualidade e funcionem como deveriam. É baseado em princípios recomendados pelo Fórum Internacional de Reguladores de Dispositivos Médicos (IMDRF), que é um padrão global.
Embora o sistema seja voluntário, vale a pena notar que a Autoridade Hospitalar, um grande comprador de dispositivos médicos para hospitais públicos, prefere significativamente produtos listados sob o MDACS. Em nossa experiência, isso muitas vezes torna a listagem um item comercial indispensável para muitos fornecedores que buscam ter sucesso neste mercado.
Classificação de Dispositivos: Nossa Compreensão dos Riscos
Quando se trata do MDACS, os dispositivos médicos são agrupados com base em quanto risco podem representar para pacientes e usuários. Essa abordagem baseada em risco, pelo que vimos, realmente molda o nível de verificações regulatórias que eles precisarão.
Dispositivos Médicos Gerais:
- Classe I (Baixo risco): Estes são dispositivos com risco mínimo. Pense em itens simples do dia a dia, como espátulas, curativos ou auxiliares de caminhada.
- Classe II (Baixo a médio risco): Esses dispositivos apresentam um pouco mais de risco. Exemplos que encontramos comumente incluem lentes de contato, aparelhos auditivos e bisturis cirúrgicos.
- Classe III (Médio a alto risco): Estes são dispositivos de maior risco. Esta categoria inclui itens mais complexos, como ventiladores, implantes de quadril e equipamentos de ultrassom diagnóstico.
- Classe IV (Alto risco): Estes são os dispositivos mais críticos, onde uma falha pode ter consequências muito sérias. Nossa experiência mostra que esta classe inclui dispositivos vitais, como marcapassos, válvulas cardíacas e desfibriladores implantáveis.
Dispositivos Médicos de Diagnóstico In Vitro (IVD):
- Classe A (Baixo risco): Estes são geralmente materiais de laboratório simples, como reagentes comuns ou meios de cultura gerais.
- Classe B (Risco moderado): Estes incluem testes para coisas como gravidez ou determinação de níveis de colesterol.
- Classe C (Alto risco): Este grupo abrange testes mais críticos, como dispositivos para autoavaliação de glicose no sangue ou detecção de agentes infecciosos como clamídia.
- Classe D (Maior risco): Estes são os IVDs mais vitais, frequentemente usados para triagens críticas de saúde pública. Com base em nosso conhecimento, isso abrange testes para detecção de agentes transmissíveis em doações de sangue (por exemplo, HIV, Hepatite C) ou para agrupamento sanguíneo.
O Processo de Listagem: Etapas que Orientamos os Clientes
Para fabricantes e importadores que buscam fornecer dispositivos médicos das Classes II/B, III/C ou IV/D em Hong Kong, obter a listagem de seus produtos sob o MDACS é, em nossa visão, um passo absolutamente essencial.
Aqui estão as etapas principais envolvidas, conforme entendemos e normalmente assistimos:
- Nomear uma Pessoa Responsável Local (LRP): Se um fabricante estiver baseado fora de Hong Kong, ele precisa absolutamente nomear um LRP. De nossa experiência, o LRP deve ser uma entidade incorporada localmente, e serve como o principal ponto de contato com o Departamento de Saúde. Essa pessoa ou entidade assume a responsabilidade significativa de lidar com a aplicação, coordenando de perto com o fabricante e cumprindo todas as responsabilidades contínuas após o produto estar no mercado.
- Preparar o Dossiê de Aplicação: Isso envolve reunir um dossiê técnico abrangente. Descobrimos que isso inclui o formulário de aplicação, evidências sólidas de um Sistema de Gestão da Qualidade certificado (como ISO 13485, que demonstra um controle de qualidade robusto), informações detalhadas sobre o dispositivo em si e documentação técnica que mostra claramente que atende aos "Princípios Essenciais de Segurança e Desempenho." De acordo com nossa experiência, ter aprovação de marketing de pelo menos uma autoridade regulatória reconhecida (como a FDA dos EUA, Organismos Notificados da UE ou Saúde Canadá) pode agilizar e acelerar significativamente o processo de revisão.
- Submissão e Revisão: O LRP submete a aplicação à Divisão de Dispositivos Médicos (MDD). A MDD então realiza uma revisão minuciosa do dossiê. Se encontrarem algo faltando ou que precise de esclarecimento, a MDD emitirá um pedido de informações adicionais. Do nosso ponto de vista, responder a essas consultas de forma rápida e abrangente é crucial para evitar atrasos.
- Listagem e Responsabilidades Pós-Mercado: Uma vez que a revisão seja bem-sucedida, o dispositivo é adicionado à lista do MDACS, e um número de listagem único é emitido. É importante lembrar que as responsabilidades do LRP continuam bem depois que o produto é listado. Isso inclui estabelecer um sistema de rastreabilidade, relatar prontamente quaisquer incidentes adversos e gerenciar quaisquer recalls ou ações corretivas de segurança em campo necessárias, como vimos acontecer no mercado.
A Importância do GN-02: Nossa Referência Principal
As "Notas de Orientação para Listagem de Dispositivos Médicos das Classes II/III/IV" (GN-02) são, do nosso ponto de vista, o guia definitivo para todo esse processo. Ele fornece detalhes muito granulares sobre os requisitos de aplicação, os vários caminhos de avaliação de conformidade (incluindo aqueles para dispositivos com e sem aprovação de país de referência anterior), necessidades de documentação precisas, especificidades de rotulagem e o formato exato para a Lista de Verificação de Conformidade dos Princípios Essenciais. Em nossa opinião profissional, uma compreensão aprofundada do GN-02 não é apenas útil; é absolutamente crítica para uma aplicação bem-sucedida.
Desenvolvimentos Futuros: O Que Antecipamos
O governo de Hong Kong está trabalhando ativamente para estabelecer um quadro legislativo mais abrangente e, eventualmente, obrigatório para dispositivos médicos. O que entendemos é que uma parte fundamental dessa estratégia é o estabelecimento do Centro de Regulamentação de Produtos Médicos de Hong Kong (CMPR). O objetivo final é afastar-se da dependência exclusiva de aprovações de países de referência em direção a uma abordagem de "avaliação primária" para novas e inovadoras tecnologias médicas. Com base em nossas percepções, o atual MDACS voluntário é uma fase de transição importante, permitindo que a indústria se alinhe a esses requisitos regulatórios propostos antes que se tornem obrigatórios.
Você tem algum dispositivo ou classificação específica em mente que poderíamos discutir mais com base nesses procedimentos?
Precisa de Ajuda Regulatória? Experimente Nossa Plataforma
Publique suas perguntas regulatórias ou solicite orçamentos de consultores farmacêuticos verificados em todo o mundo. Conecte-se com especialistas do seu mercado.
Preços Imbatíveis
Taxas transparentes e competitivas para registro de dispositivos médicos em Hong Kong.
Farmacêutico registrado · Engenheiro de IA · Diretor, ElendiLabs
Farmacêutico registrado, engenheiro de IA, fundador da HKHAIS e especialista em SEO/GEO para o domínio farmacêutico e de dispositivos médicos.
Pergunte Qualquer Coisa
Entraremos em contato pessoalmente.
100% de taxa de resposta • Resposta em até 7 dias úteis
Precisa de Orientação Especializada?
Explore Nosso Guia Interativo de Dispositivos Médicos
Obtenha orientação passo a passo sobre as regulamentações de dispositivos médicos de Hong Kong, classificação de dispositivos e requisitos de conformidade.
Ver Guia de Dispositivos MédicosEntre em contato conosco em contact@elendilabs.com / +852 4416 5550